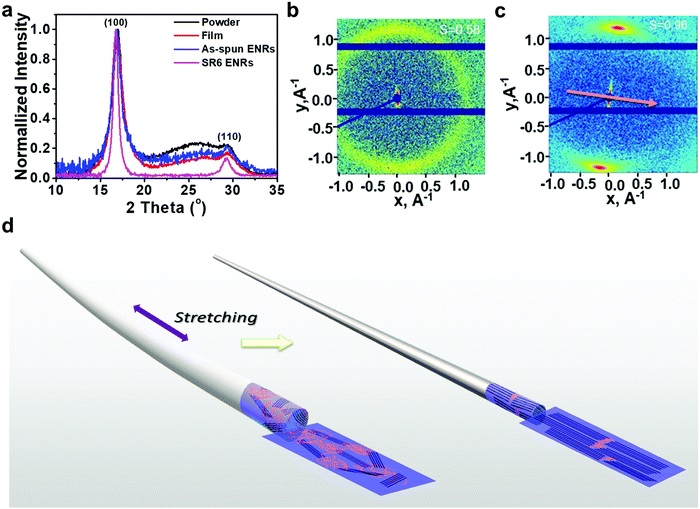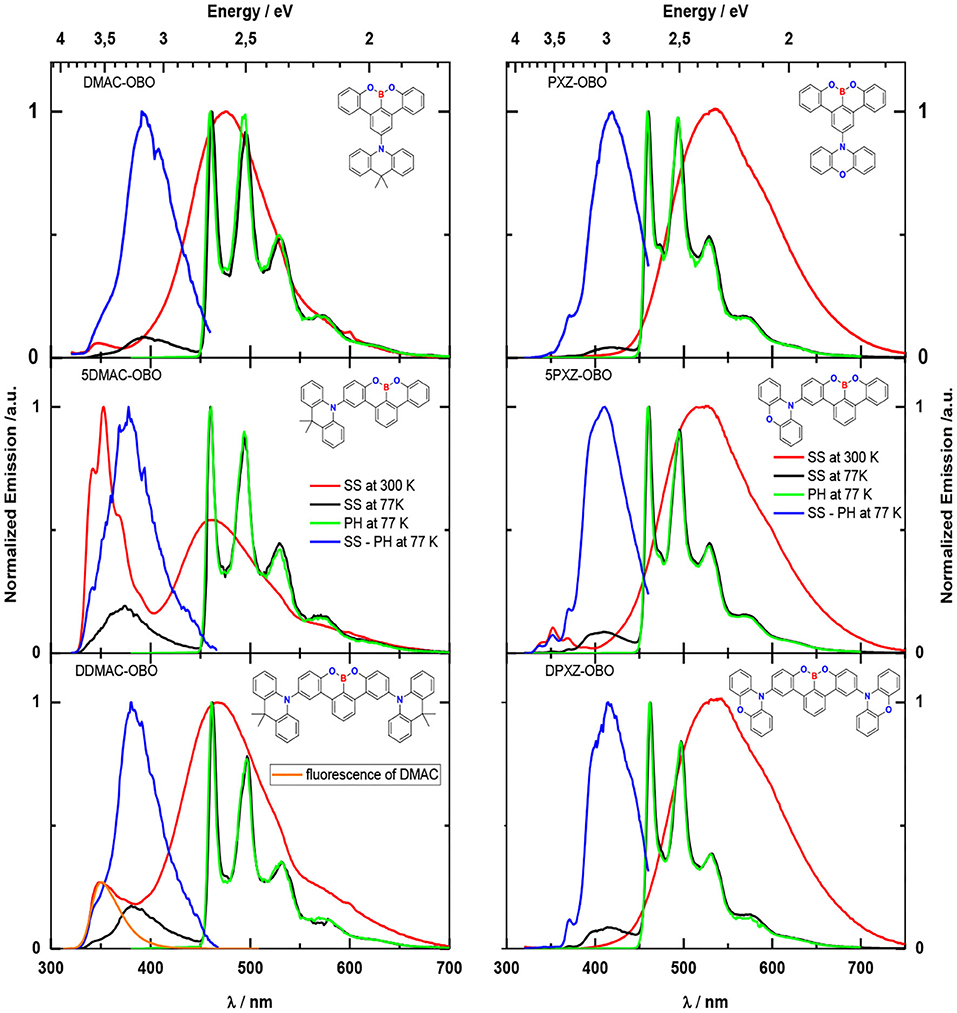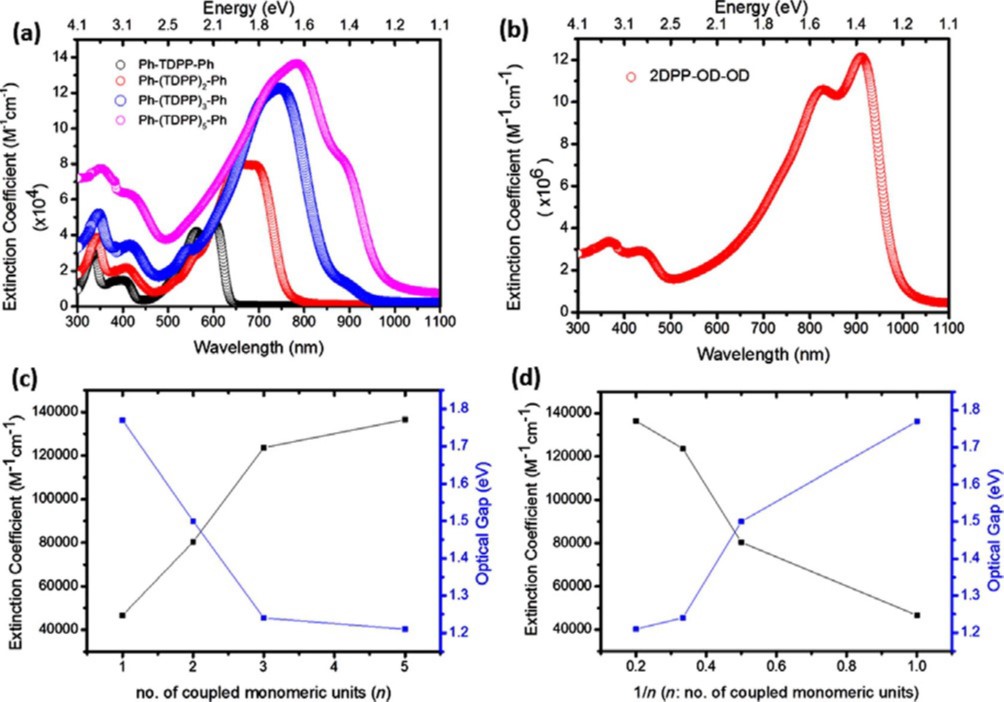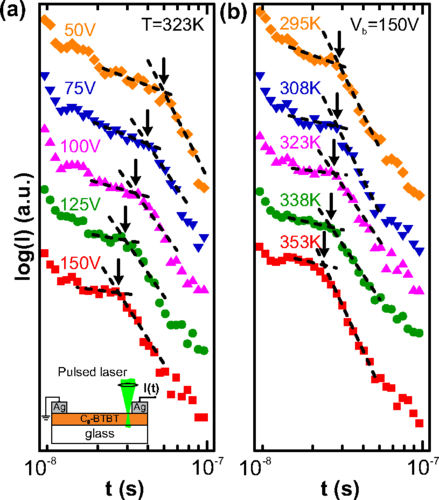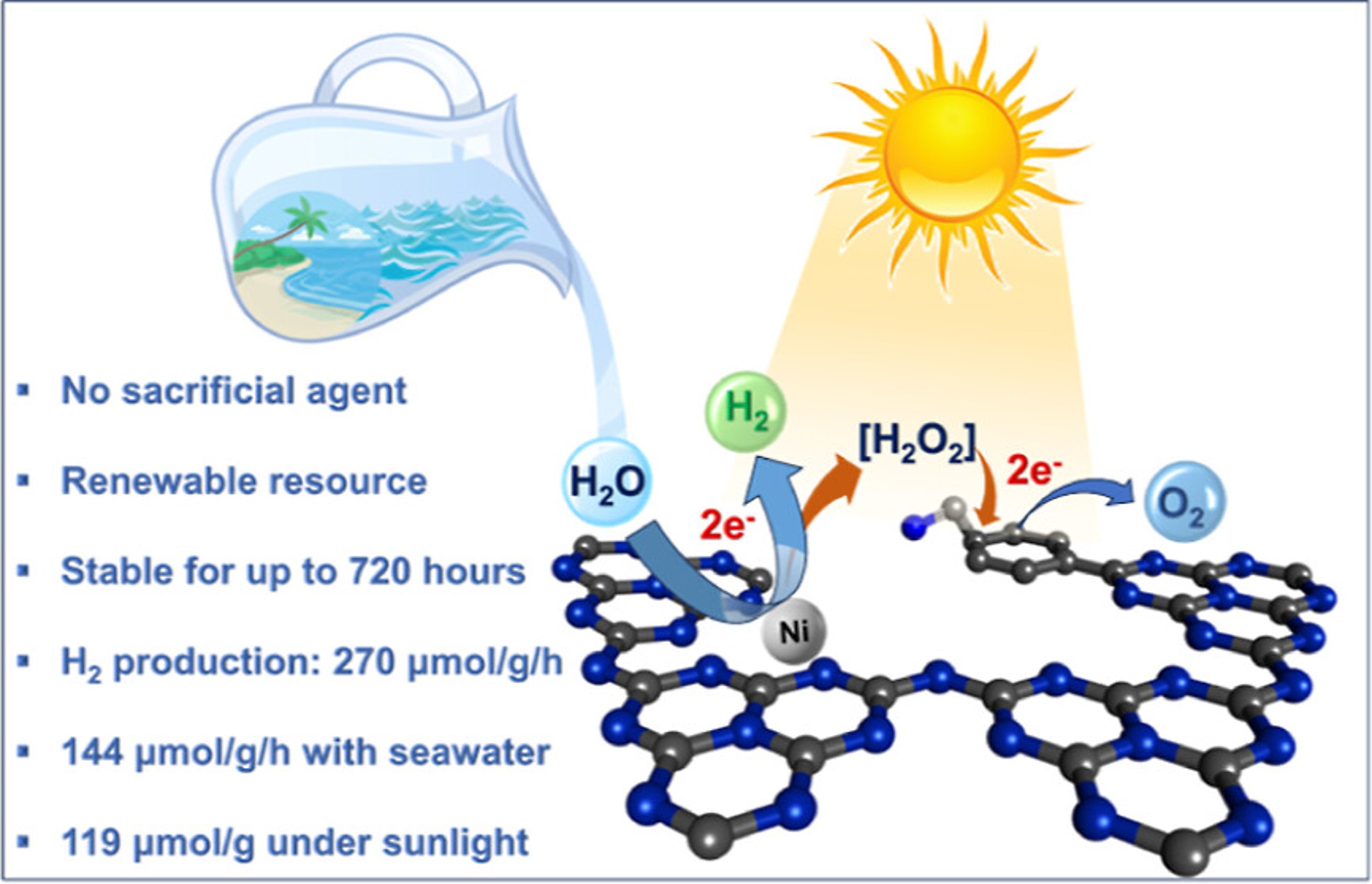
Green hydrogen is widely regarded as a key to a sustainable future, offering a clean and flexible fuel option for decarbonizing the energy, transport, and industrial sectors. While photocatalytic approaches are known for generating hydrogen directly from water, most existing methods require (over)stoichiometric amounts of sacrificial reagents, which is far from ideal for the production of green hydrogen. To address this challenge, we have developed an atomically dispersed Ni-based photocatalyst that achieves hydrogen evolution rates of up to 270 μmol/g/h (168 mmol/gNi/h). Remarkably, this photocatalyst also exhibits high photoreactivity under direct sunlight, producing up to 17 μmol/g/h (10.6 mmol/gNi/h) of hydrogen. Impressively, the catalyst can even generate green hydrogen directly from seawater, up to 144 μmol/g/h, demonstrating significant potential for real-world applications. The photocatalyst is exceptionally stable, remaining active even after 720 h (140 h of irradiation and 580 h resting time) of operation and retaining high performance over more than 15 cycles. Furthermore, comprehensive spectroscopic and structural analyses─including HRTEM, PXRD, ssNMR, XPS, and XAS─provide detailed structural insights and confirm the atomically dispersed nature of the Ni species. In-depth mechanistic studies have elucidated the critical role of atomic dispersion in enabling robust photocatalytic efficiency.